
Best practice for crystallisation kinetics measurements via NMR
- Like
- Digg
- Del
- Tumblr
- VKontakte
- Buffer
- Love This
- Odnoklassniki
- Meneame
- Blogger
- Amazon
- Yahoo Mail
- Gmail
- AOL
- Newsvine
- HackerNews
- Evernote
- MySpace
- Mail.ru
- Viadeo
- Line
- Comments
- Yummly
- SMS
- Viber
- Telegram
- Subscribe
- Skype
- Facebook Messenger
- Kakao
- LiveJournal
- Yammer
- Edgar
- Fintel
- Mix
- Instapaper
- Copy Link
Posted: 2 April 2017 | Gottfried Ziegleder - Fraunhofer Institute for Process Engineering and Packaging, Isabell Rothkopf - Fraunhofer Institute for Process Engineering and Packaging, Wolfgang Danzl - Fraunhofer Institute for Process Engineering and Packaging | No comments yet
Low resolution or pulsed NMR (nuclear magnetic resonance) is an easy and fast way to determine solid fat content (SFC) of cocoa butter and other fats. It can also be used to observe the crystallisation process. However, in literature several ways of sample preparation and handling, especially for crystallisation kinetics measurement, are reported. These differences in preparation might not be critical when comparing results from one laboratory, but could be crucial when interlaboratory results are compared. The latter one is the case when it comes to quality control of raw materials. In this study different sample preparation and handling methods were used and their impact on the final results was investigated.

Today, in most cases NMR is used for standardised solid fat content (SFC) measurement.1,2 However, measuring crystallisation and melting processes of cocoa butter was already described in 1980.3 Ever since NMR was used to obtain crystallisation kinetics, but sample preparation and measuring conditions varied considerably.4 In this study we compare proceedings described in literature and hand out advice on best practice. To facilitate the evaluation of influencing factors, standardised SFC measurements were used. Each measurement consisted of three scans with 2s recycle delay. Temperature was recorded using a thermologer Almemo 8590-9. For temperature measurement inside the NMR probe head fibre optics were used.
For SFC measurements a direct5 or an indirect6 method can be used.4 The direct method is used for pure fat samples without particles, such as cocoa butter, while the indirect method is suitable for samples containing particles, such as chocolate or cocoa nibs, liquor and powder.4
Sample preparation
A difference between the direct and the indirect method, which will be in focus at the end of the article, is the filling height of the samples. Samples are
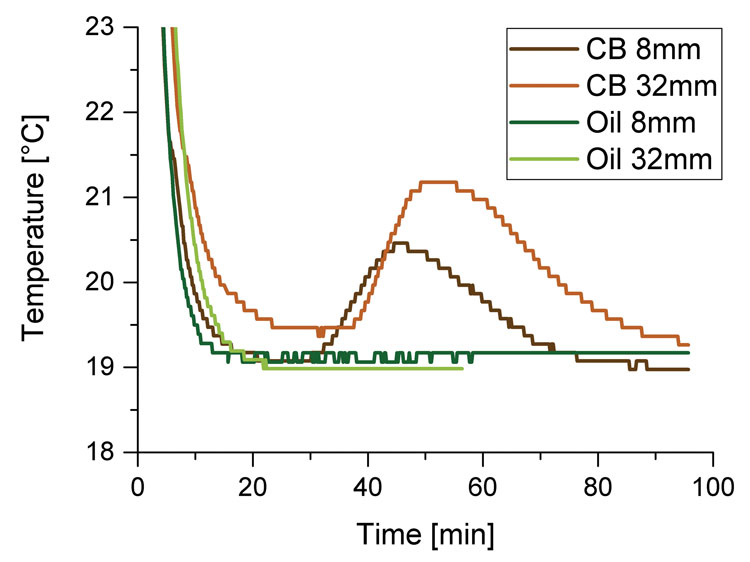
Figure 1: Temperature in cocoa butter (CB) and oil samples with a filling height of 8 and 32 mm in a NMR probe head tempered to 19 °C
placed in glass tubes with 10mm diameter. For measurements using the direct method, filling height is 30‑40mm; for indirect method it is 8‑10mm.4 This means that the volume of samples that needs to be cooled is four times smaller in case of the indirect method compared to the volume used for direct method measurements. For the smaller filling height the spacer was used. The heat release of cocoa butter and vegetable oil samples with eight and 32mm filling height in a tempered NMR probe head can be seen in Figure 1.
Due to the lesser warming of the sample with smaller filling height, we suggest the indirect method for crystallisation kinetic measurements.
Sample handling
Before each measurement samples were fully melted and homogenised before placing the respective amount in an NMR tube. Prior to the measurement, the sample was heated to 70°C for 30min to erase thermal history. Afterwards crystallisation is initiated by putting the sample into an aluminum block, water bath or in the tempered NMR-probe head. In doing so, the sample might be rocked. Rocking might disturb the arrangement of the molecules or it might initiate the crystallisation especially in pure cocoa butter samples. Keeping your sample steady is recommended.
Choosing a crystallisation temperature
The temperature of the probe-head, which is the measuring temperature, is often chosen in accordance to DSC measurements. However, it could also
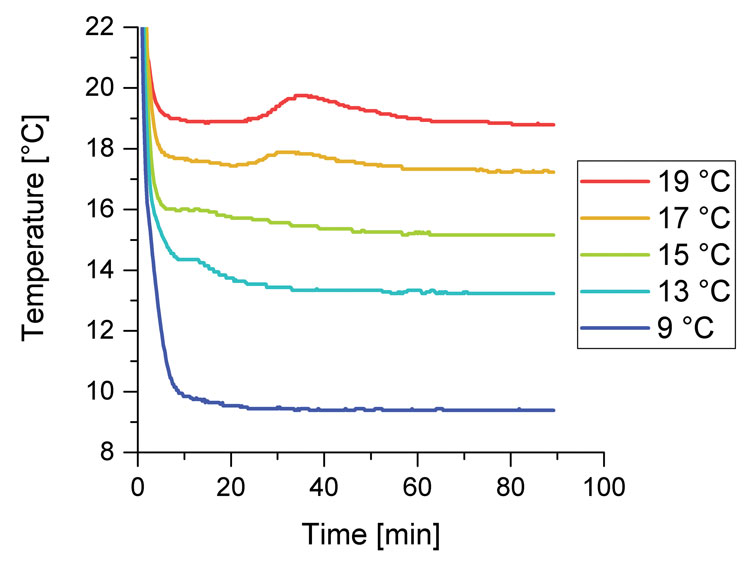
Figure 2: Temperature progression in cocoa butter samples (filling height: 8 mm) cooled in an aluminum block at different temperatures
be in the range of MultiTherm or Shukoff measurements, which would be at 17.6°C and 0°C or 8°C, respectively.7,8 Therefore the temperature progression in an NMR tube with cocoa butter at a filling height of 8mm was measured for different given temperatures of an aluminium block in a water bath, shown in Figure 2.
All curves show a temperature decrease in the first 5-10min. The release of latent crystallisation heat starts earlier for lower given temperatures. At 19, 18 and 17 °C the temperatures decrease in the beginning and the increase during crystallisation are baseline separated. This is not the case for lower temperatures.
Sample tempering
Sample tempering can be done in a water bath, in an external aluminium block or inside the NMR. The heat release in a water bath is better due to
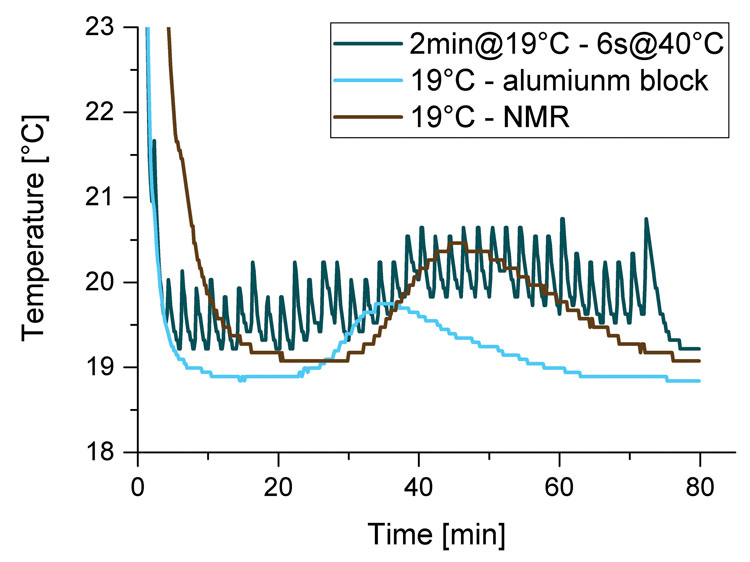
Figure 3: Temperature progression in a cocoa butter sample cooled to 19 °C in the NMR probe head, in an aluminum block and with simulated measurement in an aluminum block at 40 °C for 6 s every 2 min
improved heat transfer in water compared to air. However, care has to be taken when wiping off the water.9 To investigate if the remaining water influences the measurement 20 samples were prepared as described for standardised SFC measurement.5 The samples were divided in two groups in which the last tempering step of 1h at 20°C varied. For one group the last step was water free in an aluminium block tempered in a water bath. The second group was placed directly in the water bath and the water was wiped off the tubes just before the measurement.
A t-test showed, that the two groups are statistically significant different with a=0.05 and P£0,001.
However, heat transfer in an aluminium block and inside the NMR probe head also differs. Therefore the temperature progression in a cocoa butter sample (filling height: 8mm) in an aluminium block and in the NMR probe head (with NMR turned off) at 19°C was compared, as can be seen in Figure 3.
The temperature equilibrates faster in the aluminium block than in the NMR probe-head. There is hardly a baseline separation in the sample cooled in the NMR probe head.
Wiping off the water from a sample tempered indirectly in a water bath is individually different and depends on the operator. Therefore tempering in an external aluminium block or directly in the NMR probe-head is recommended. External tempering allows performing several measurements at the same time by hand or by using an auto sampler. However, when the sample is moved by hand, rocking and shaking should be avoided. Internal tempering can be used with automated measurement, but for only one sample at a time.
Probe-head tempering during the measurement
During measurements with external tempering, the NMR probe-head can optionally be tempered to the crystallising temperature too. The use of an external tempering unit without tempering the probe head was simulated by manually putting a molten sample (cocoa butter with filling height of 8mm) in an aluminium block cooled to 19°C for 2min followed by 6s in an aluminium block at 40°C. This procedure was repeated until the latent heat release during crystallisation could no longer be observed. The temperature progression of a cocoa butter sample kept in the aluminium block at 19°C and a sample with simulated measurement for 6s at 40°C is shown in .
The results show, that even during the short measuring time, the sample warms up. Temperature increases up to 2°C and thus increases the effective crystallisation temperature and extends the heat release of the sample. Therefore tempering the NMR probe-head to crystallisation temperature is always recommended.
Choosing your method: direct vs. indirect
For the direct method5 only one measurement is needed. The ratio of the signal in the beginning, including the solid and the liquid phase, and the
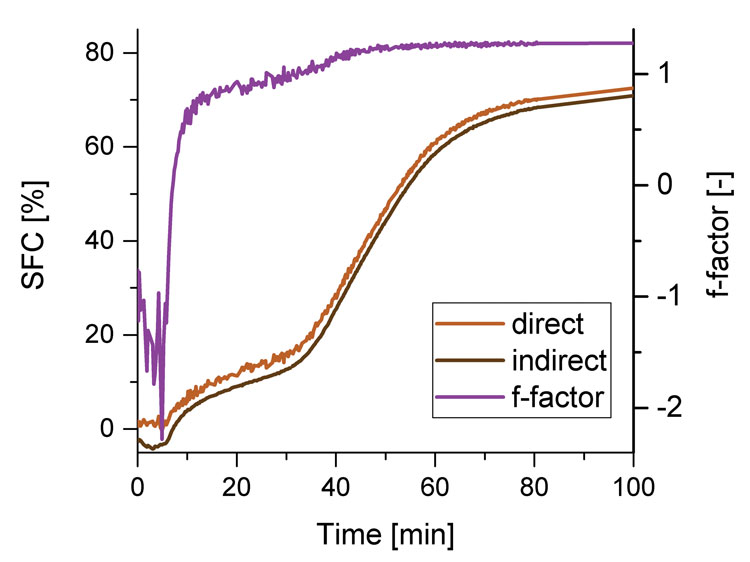
Figure 4: SFCindirect, SFCdirect and f-factor during crystallisation measurement
second signal, which includes the signal of the liquid phase is used to calculate the SFC. Due to the dead time, the first signal of the solid and liquid phase cannot be detected directly after the high frequency impulse. An extrapolation factor f is used to calculate the signal at time t=0. Therefore the direct method results in an approximated value because of the f-factor for several reasons. First, a linear extrapolation is assumed to calculate the signal at t=0, but the signal shows a non-linear decrease. Additionally, the f–factor depends on the molecular mobility, e.g. temperature, polymorphism and crystal size.10,11 Additionally its temperature dependency is also due to the dilatation of the liquid phase. Since the f-factor depends on the polymorphic state, it changes during the crystallisation process, which can be seen in Figure 4. From the recorded signals, the direct SFC, the indirect SFC and the f-factor were calculated as described in.5,6
Another influencing value is the solid phase in samples with non-fat solids. Therefore two measurements of a sample, one at the solidified and one at melted state are necessary to distinguish between protons in the crystallised fat and those in the solid non-fat material of chocolate (non-fat cocoa solids, sugar). Ziegleder et al. (1998) described this problem and developed a correction function to calculate the SFC of chocolate.12
To avoid these insecurities, we recommend the indirect method. For the indirect method, only the signal of the liquid phase is used.6 The signal of the molten and the crystallised sample, as well as the signal of a reference at the same temperature is measured. The values of the reference are needed for temperature correction. As reference oils, which are liquid at the measuring temperature range, e.g. sunflower, canola or hazelnut oil can be used. Additionally, online recording of the temperature throughout the measurement would allow considering the temperature increase during crystallisation.
Acknowledgment
The authors thank Christian Schormair for conducting the lab work. This project of precompetitive research was supported by the Chocolate Group of the Industrial Association of Food Technology and Packaging e.V. (IVLV) Freising.
References
- Leung HK, Anderson GR, Norr PJ. Rapid Determination of Total and Solid Fat Contents in Chocolate Products by Pulsed Nuclear Magnetic Resonance. J Food Science 1985;50:942–45; http://dx.doi.org/10.1111/j.1365-2621.1985.tb12985.x.
- Petersson B, Anjou K, Sandström L. Pulsed NMR Method for Solid Fat Content Determination in Tempering Fats, Part I: Cocoa Butters and Equivalents. Fette, Seifen, Anstrichm. 1985;87:225–30; http://dx.doi.org/10.1002/lipi.19850870603.
- Brosio E, Conti F, Nola A, Sykora S. A pulsed low resolution NMR study on crystallization and melting processes of cocoa butter. J Am Oil Chem Soc 1980;57:78–82; http://dx.doi.org/10.1007/BF02674367.
- Padar S, Jeelani SAK, Windhab EJ. Crystallization Kinetics of Cocoa Fat Systems: Experiments and Modeling. J Am Oil Chem Soc 2008;85:1115–26; http://dx.doi.org/10.1007/s11746-008-1312-0.
- Solid fat content (SFC) by low-resolution nuclear magnetic resonance—the direct method.
- Solid fat content (SFC) by low-resolution nuclear magnetic resonance—the indirect method.
- Shukoff AA. Über eine neue Methode zur Bestimmung der Erstarrungstemperatur. Z. Angew. Chem. 1899;12:563–64; http://dx.doi.org/10.1002/ange.18990122403.
- Bühler. Bedieungsanleitung MultiTherm. Uzwil.
- Marangoni AG, Peyronel MF. Pulsed Nuclear Magnetic Resonance Spectrometry: AOCS; 2014.
- Trezza E, Haiduc AM, Goudappel GJW, van Duynhoven JPM. Rapid phase-compositional assessment of lipid-based food products by time domain NMR. Magn Reson Chem 2006;44:1023–30; PMID:16953523; http://dx.doi.org/10.1002/mrc.1893.
- Adam-Berret M, Mariette F. New developments in low field NMR for the characterisation of food microstructure. New Food 2009.
- Ziegleder G, Schwingshandl I, Brulheide M. Beurteilung gelagerter Schokoladen anhand NMR-Kernspinresonanz. Süßwaren 1998:30–34.
Issue
Related topics
Related organisations
Industrial Association of Food Technology and Packaging (IVLV)





