
Microbial metagenomics and the food industry
- Like
- Digg
- Del
- Tumblr
- VKontakte
- Buffer
- Love This
- Odnoklassniki
- Meneame
- Blogger
- Amazon
- Yahoo Mail
- Gmail
- AOL
- Newsvine
- HackerNews
- Evernote
- MySpace
- Mail.ru
- Viadeo
- Line
- Comments
- Yummly
- SMS
- Viber
- Telegram
- Subscribe
- Skype
- Facebook Messenger
- Kakao
- LiveJournal
- Yammer
- Edgar
- Fintel
- Mix
- Instapaper
- Copy Link
Posted: 17 January 2017 | Greg Jones, Campden BRI | No comments yet
In New Food’s latest coverage on the topic of what genomics can do for the food industry, we explore insight from Greg Jones at Campden BRI and his expertise on microbial metagenomics…

In New Food’s latest coverage on the topic of what genomics can do for the food industry, we explore insight from Greg Jones at Campden BRI and his expertise on microbial metagenomics…

Imagine a world in which microbial populations are determined without needing to grow them on agar. Imagine being able to find organisms that we cannot culture in the lab. Imagine being able to pick up not just the healthy members of a community, but those that are injured as well. This is now a reality through a new technique known as ‘metagenomics’.
The potential applications for this technique are limited only by the populations we can find, making it ideal for spoilage investigations, shelf life analyses and environmental monitoring (to name but a few!). It will also provide more information to the food manufacturer used to a simple count, as this count will now be much more closely related to the species mix in the product. The following article aims to introduce the reader to this exciting new field of microbial analysis.
Metagenomics is a technique that takes advantage of recent advances in sequencing technology allowing huge numbers of different individual DNA sequences to be read at any one time. This means a heterogeneous population of DNA molecules can be read directly from a DNA extract. The advantages of this are that previously uncultured organisms can be identified and little manipulation of is needed in order to produce data regarding the composition of that sample which reduces experimental biases.
This works best for samples which are expected to contain more than one species of organism, and thus it is currently used to analyse food mixes (e.g. meat mixtures or herb & spice mixes) to determine authenticity, and microbial populations to analyse spoilage organisms. An example of the output of a metagenomic analysis of the microbial population of a poultry sample is shown below:
Figure 1: Krona chart of the microbial population of a poultry sample as determined via sequencing a pool of 16S ribosome gene amplicons.
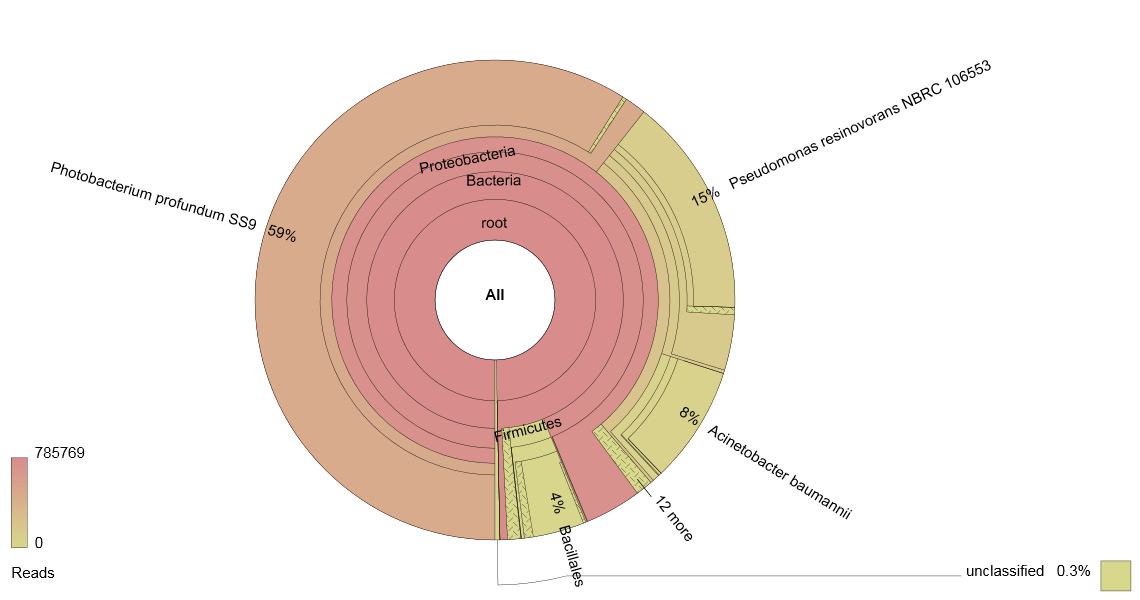
Metagenomics is only as good as the database against which the sequences are compared, and this fact presents a dilemma. DNA extracted directly from a product will be more representative of the original sample than if any further manipulation has been carried out. The resulting sequences will comprise a random sub-set of the total population, and it is possible that these will not be held in a database with an accompanying identifier. Therefore, whilst being more representative of the overall population, the chance of much of one’s data residing in the ‘Unclassified’ section of an analysis increases.
To increase the chances of identification, PCR can be used to generate a pool of sequences for which there may be better database support. In microbiology, the gene chosen in the overwhelming majority of cases is the gene coding for the 16S sub-unit of the ribosome (16S rDNA). This gene is used for the majority of microbial taxonomy studies and has extensive database support. Running a PCR has the advantage of ‘cleaning’ the sample of sequences that are not 16S rDNA sequences (e.g. DNA from the host food), however the choice of primers will exclude some organisms, primarily fungi, from the analysis. This can be addressed by running a separate analysis using fungi-specific primers.
The ‘Holy Grail’ of microbial Metagenomics is to replace culture-based methodology for enumeration…
In Figure 1, a 16S rDNA profile has been generated for a spoiled poultry sample. If a culture-based analysis had been used for this sample, the investigator would have chosen the media to be used to isolate various groups of organisms a priori. In this case, it is likely that Pseudomonas spp., Enterobacteriaceae and a Total Viable Count (TVC) would feature in the analysis. The results in Figure 1 give an indication of the influence these choices can have on understanding the microflora of a product.
Instead of the accepted wisdom that the dominant microflora would be comprised of Pseudomonas spp., the highest proportion of reads (59%) is assigned to Photobacterium profundum, an organism currently associated with fish. Enterobacteriaceae are not shown to be significant. TVC and Pseudomonas spp. counts potentially offer less information about the microflora of the sample than this 16S rDNA amplicon analysis.
The ‘Holy Grail’ of microbial metagenomics is to replace culture-based methodology for enumeration. The 16S rDNA amplification currently represents the most accepted way of generating a microbial profile, despite the risk of PCR amplification introducing bias. The relationship between the profile and the number of cells in the sample will take much more study, however initial results show that similar general trends can be observed. In January 2017 Campden BRI started a 3 Year Member-funded project designed to use microbial profiling to better investigate the microflora of a range of food types over their shelf lives.
______________________________________________________________________
This data will be compared against culture based methodology to highlight discrepancies and compare the trends observed. If you are interested in being a part of this project, please get in touch: [email protected] or 01386 842143.




